
2024 brings great news! The fourth high-quality article on the FAD4T Alzheimer's disease mouse model, developed independently by GemPharmatech, has been published!

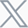
On January 23, 2024, the research team of Chen Ming from Anhui Medical University and the research team of Tang Binliang from Zhejiang University School of Medicine collaborated with scientists from GemPharmatech to publish online in the Life Sciences (IF = 6.78) journal a paper titled "Cognitive impairment in Alzheimer's disease FAD4T mouse model: Synaptic loss facilitated by activated microglia via C1qA." This study used the FAD4T (C57BL/6JGpt-Tg (Thy-APP/Thy-PSEN1) 5/Gpt, T053302) mouse model, which is independently developed by GemPharmatech, as a model for cognitive impairment in Alzheimer's disease (AD), and discovered a series of molecular changes related to cognitive dysfunction. Through behavioral tests and immunofluorescence staining techniques, the researchers observed the presence of Aβ plaques in the brains of FAD4T mice as well as abnormal activation of microglia in the cortex and hippocampus, further confirming the occurrence of cognitive impairment. Using patch clamp recordings of hippocampal neurons and Golgi staining techniques, the researchers found a decrease in the amplitude of postsynaptic currents and abnormal synaptic morphology, indicating synaptic dysfunction in the FAD4T mouse model. Further RNA-seq analysis revealed increased expression of immune system and pro-inflammatory genes, particularly an increase in C1qA protein and mRNA levels, suggesting that over-activation of microglia may be a key factor leading to synaptic loss in neurons. Overall, this study provides a comprehensive understanding of the mechanism of cognitive impairment in AD patients and preliminarily explores potential therapeutic approaches targeting C1qA. This research is expected to provide important guidance for the development of more effective treatment strategies in the future and offer a new perspective for better understanding and addressing the challenging disease of AD. [1]

Previous studies have shown that excessive activation of glial cells, especially the microglia and astrocytes surrounding Aβ plaques, leads to neuronal apoptosis, resulting in memory loss and cognitive dysfunction [2,3]. In addition, microglial phagocytosis plays a crucial role in synaptic loss [4,5,6]. Microglia fine-tune neural circuits by engulfing excessive synapses, a process that is partially dependent on complement-mediated synaptic pruning. Specifically, as an initiator of the classical complement pathway, C1q is produced by microglia in the brain, co-localizes with synaptic markers, and may contribute to synaptic loss in various neurodegenerative models [7,8,9]. The mechanism of synaptic loss induced by complement activity still requires further investigation. Therefore, this study further investigated this mechanism at different levels using one of the AD models - the FAD4T mouse model.
After first establishing the pathological phenotype and behavioral impairments of FAD4T mice, the authors explored the density of dendritic spines and the expression of postsynaptic scaffold protein PSD-95 using Golgi staining and Western blot, respectively. The results showed a significant decrease in dendritic spine density in the cortex and hippocampus of FAD4T mice (Figure 1 A-F), as well as a significant reduction in PSD-95 expression in the hippocampus (Figure 1 G-H). Dendritic spines, as crucial structures for receiving presynaptic signals, are essential for synaptic function in terms of their number and morphology. Subsequently, the authors recorded changes in postsynaptic currents using patch-clamp recording. The results revealed a significant decrease in the amplitude of hippocampal mEPSC in FAD4T mice compared to the control group, indicating impaired excitatory synaptic transmission in this mouse model (Figure 2 A-F). Furthermore, the authors quantified the expression of excitatory postsynaptic receptors and found a significant reduction in the NMDA receptor subunit NMDAR1, which mediates excitatory postsynaptic function, in the hippocampus of FAD4T mice (Figure 2 G-J). These results collectively indicate abnormal postsynaptic function in FAD4T mice.
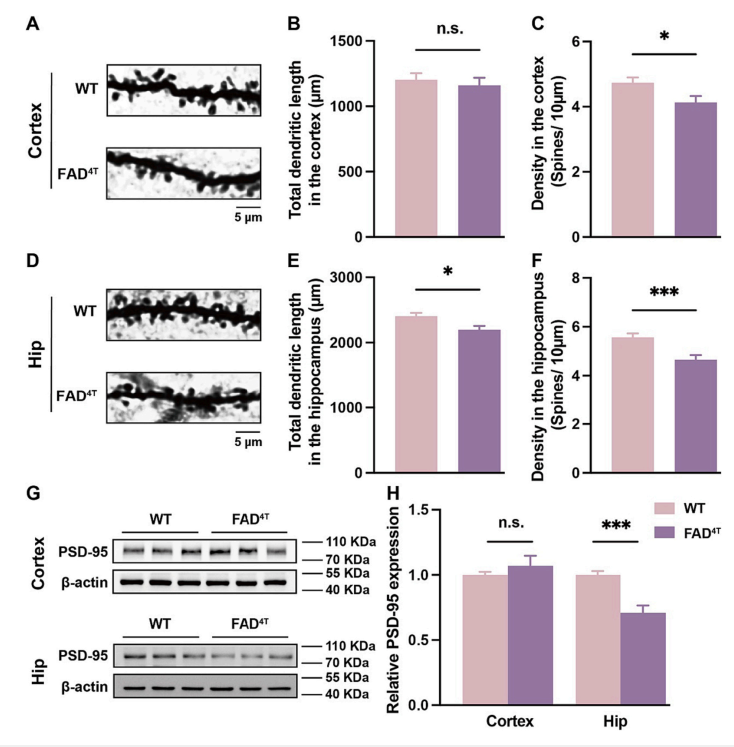
Figure 1. FAD4T mice showed abnormal synaptic loss.
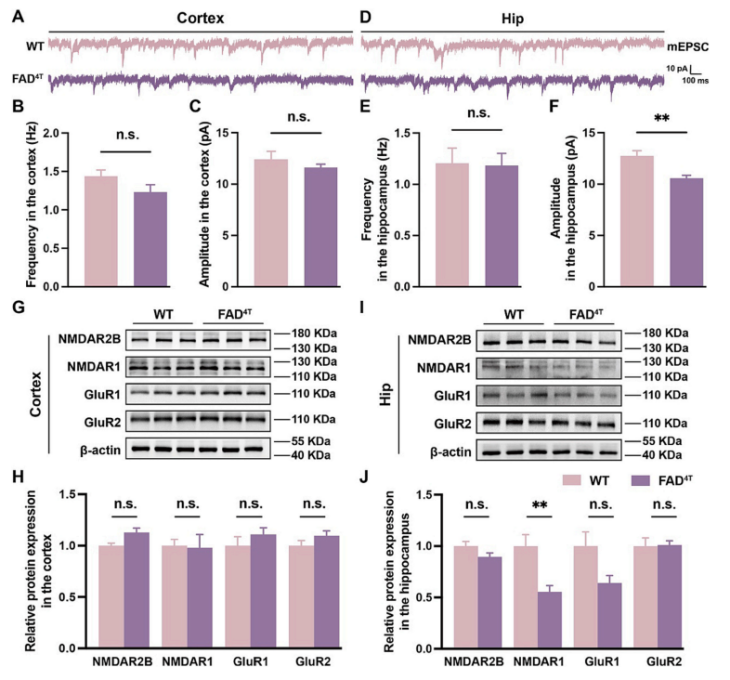
Figure 2. Reduced mEPSC amplitude and excitatory postsynaptic receptors in the hippocampus of FAD4T mice.
To further explore the potential pathogenic mechanisms underlying cognitive dysfunction in FAD4T mice, the authors performed RNA-seq analysis on the cortex and hippocampus of WT and FAD4T mice. The results showed a large number of differentially expressed genes (DEGs) in the hippocampus and cortex of FAD4T mice. Many immune system-related genes (such as Ccl4 and Ccl6) and genes highly associated with AD pathogenesis and glial cell activation (such as Gfap, CD68, cst7, and Trem2) were upregulated. Additionally, complement factor C1qA was significantly upregulated in both tissues (Figure 3 A-E), and the products of these DEGs interacted with each other in a protein-protein interaction network (Figure 3F-G). These results collectively indicate that the immune system, including the complement system, in microglia is activated in FAD4T mice, which is also considered an important factor in the pathogenesis and development of AD.
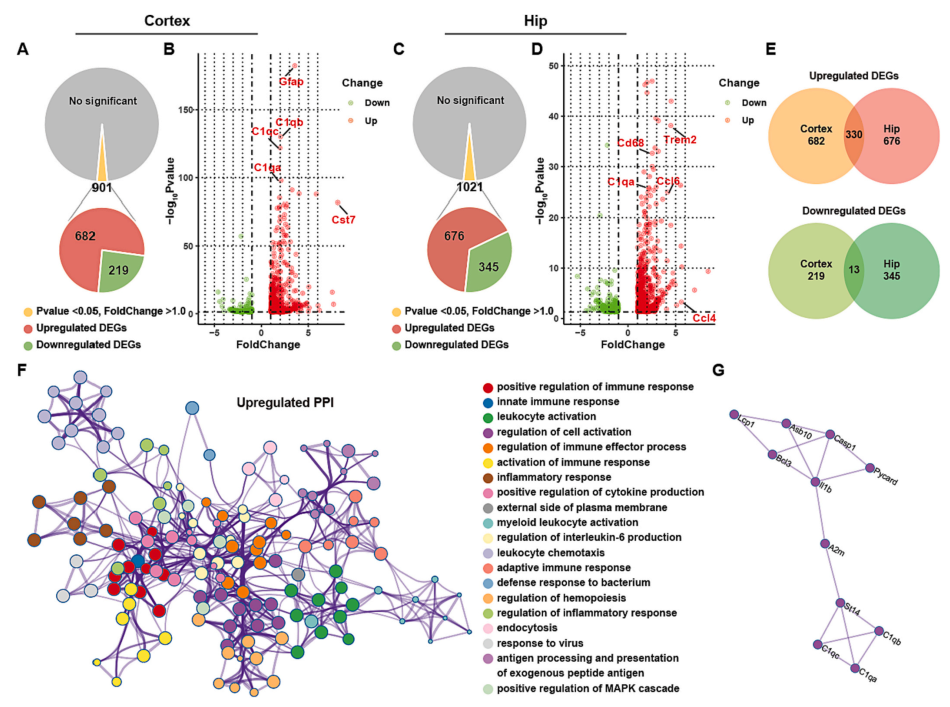
Figure 3. Differentially expressed genes in FAD4T mice in RNA sequencing results.
C1q is a component of the classical complement pathway mainly expressed in microglia, and C1qA, as one of its members, is associated with various neurological disorders. Therefore, the authors further investigated the potential impact of C1qA on neuroinflammation and synaptic function in FAD4T mice. Results from WB and qPCR showed a significant increase in C1qA protein and mRNA levels in the cortex and hippocampus of FAD4T mice (Figure 4A-F). Immunofluorescence staining also revealed a significant elevation in C1qA concentration in the surrounding activated microglia in both cortex and hippocampus of FAD4T mice (Figure 4 G-J). Furthermore, compared to WT mice, the protein and mRNA expression levels of inflammatory factors - TNF-a and IL-18 - were significantly elevated in the cortex and hippocampus of FAD4T mice (Figure 4 A-F). These results suggest that the complement component C1qA contributes to the exacerbation of neuroinflammation in FAD4T mice.
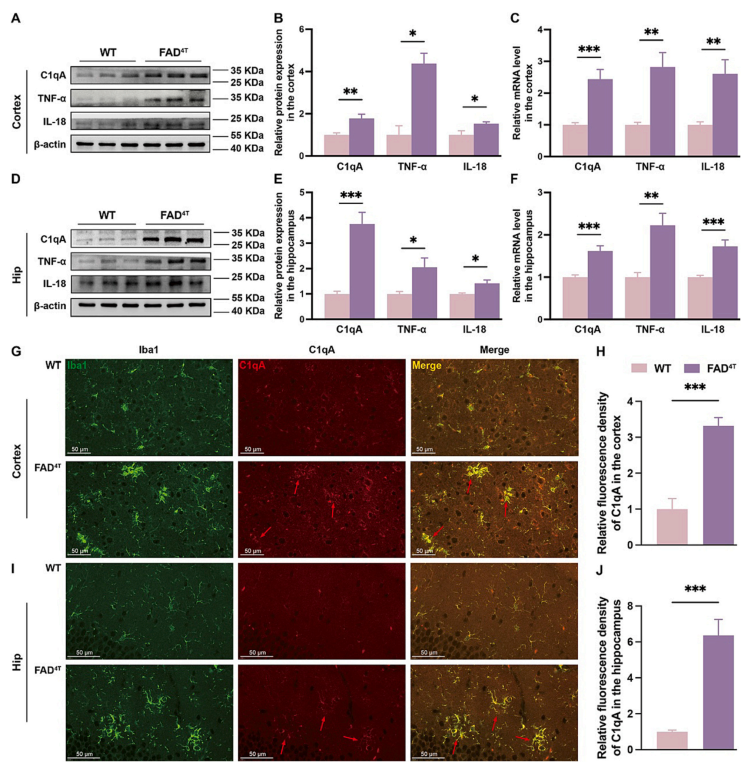
Figure 4. Increased expression levels of inflammatory factor TNF-α, IL-18 and complement component C1qA of FAD4T mice.
Studies have shown that oxytocin (OT), as a hormone peptide primarily responsible for parturition, bonding, and lactation, is also involved in regulating cognitive behaviors in the central nervous system of rodents. Therefore, the authors next investigated whether intranasal administration of OT could rescue the impaired learning and memory function in the FAD4T mouse model. Seven weeks after administration, results from the Morris water maze test showed that the escape latency of FAD4T mice was reduced after OT treatment, with an increase in the number of platform crossings and total crossings, indicating that OT can improve cognitive deficits in FAD4T mice (Figure 5 A-H). Immunofluorescence results demonstrated that the size and number of Aβ plaques in the hippocampus were reduced, along with a significant decrease in the number of microglia in the OT-treated FAD4T mice (Figure 6 A-C). Additionally, quantitative studies on complement component C1qA and inflammatory factors TNF-α, IL-1β, and IL-18 showed that OT treatment significantly reduced the expression of C1qA and inflammation-related components in FAD4T mice (Figure 6 D-J). These results collectively suggest that OT may reduce neuroinflammation levels and alleviate synaptic defects by lowering the expression of C1qA and reducing the production of Aβ plaques, ultimately leading to an improvement in cognitive deficits in FAD4T mice.
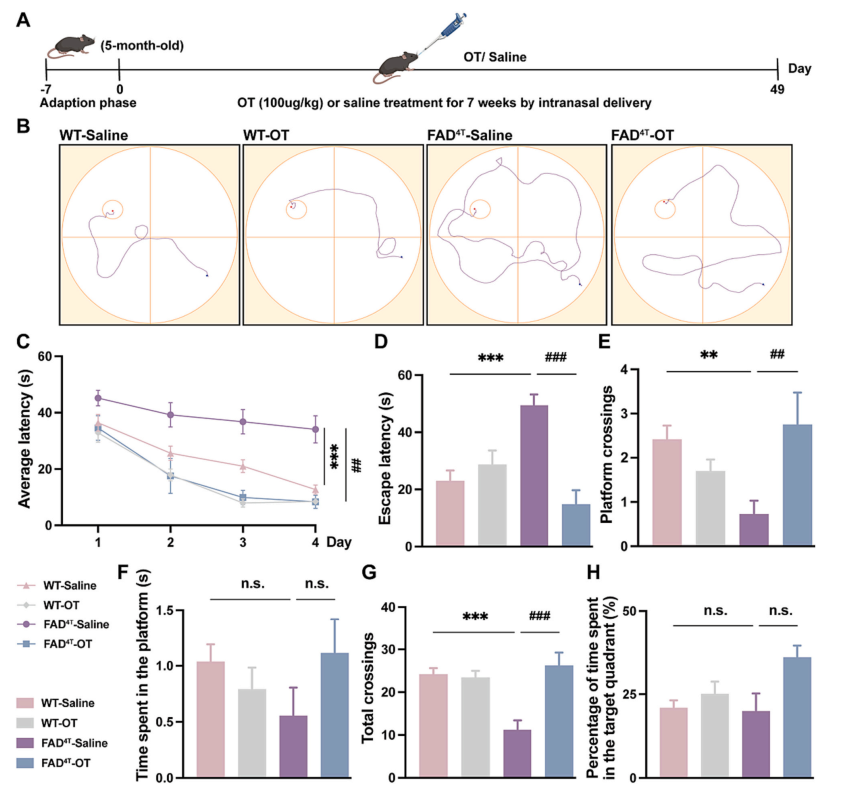
Figure 5. OT treatment improves cognitive deficits in FAD4T mice
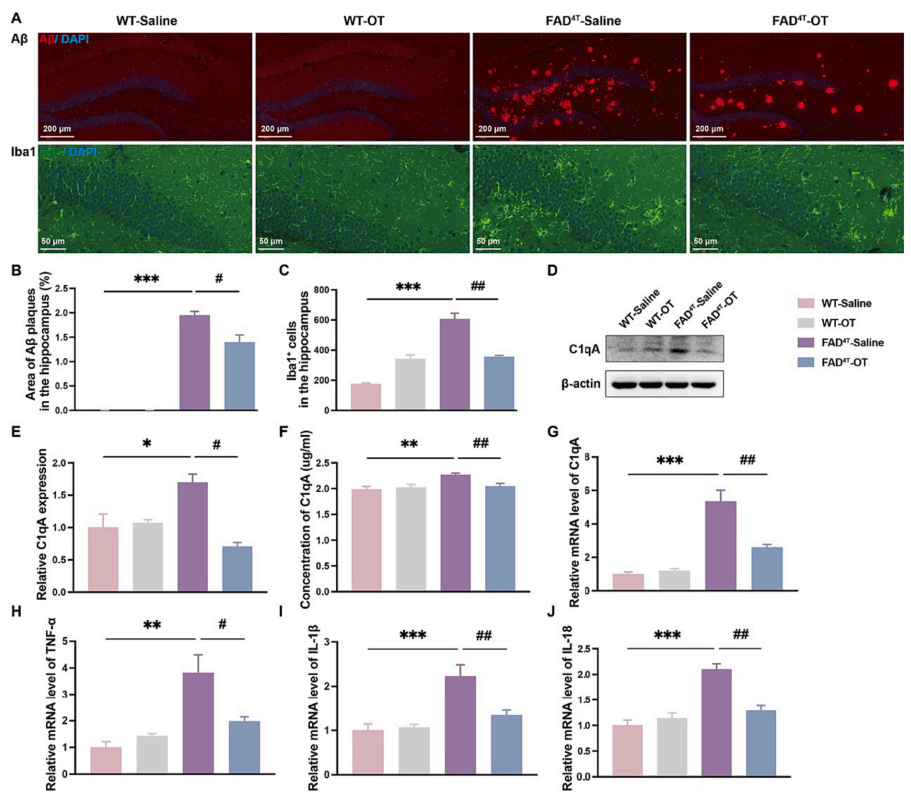
Figure 6. OT reduces the number of Aβ plaque and microglia and alleviates neuroinflammation via C1qA in FAD4T mice.
In summary, this study utilized FAD4T mice as an AD model and found that: 1) FAD4T mice exhibit cognitive impairments along with mild social deficits; 2) FAD4T mice show phenotypes such as Aβ plaque deposition, neuroglial cell activation, and upregulation of immune activity; 3) FAD4T mice display abnormal synaptic loss and synaptic dysfunction; 4) Upregulation of the complement component C1qA mediates neuroinflammation in FAD4T mice; 5) OT may improve spatial memory deficits in AD mice by potentially inhibiting C1qA-dependent neuroinflammation (Figure 7).
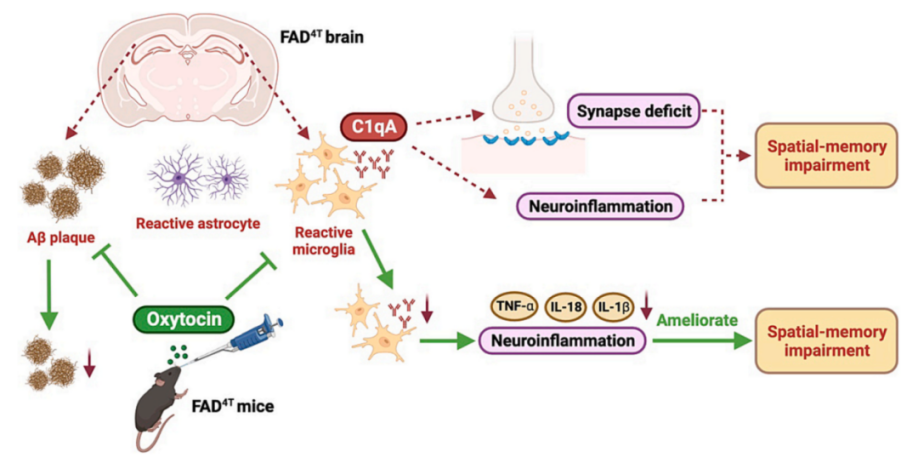
Figure 7. A working model shows the mechanism underlying OT regulation of C1qA mediated synaptic function and spatial memory in FAD4T mice.
The self-developed FAD4T mouse model of GemPharmatech, which has the humanized Swedish and Indiana mutations in the APP gene and the humanized M146L and L286V mutations in the PSEN1 gene, can effectively simulate the disease characteristics of clinical AD patients and meet the related efficacy requirements. Currently, four related articles have been published, and more related articles based on this model are either under submission or review, and it is believed that more high-quality articles will be available in the future. In addition to this model, GemPharmatech also offers the FAD3T, FAD2T, and AD Plus mouse models carrying humanized targets (such as hTREM2) based on the FAD4T model. For more details about these models, please feel free to contact your familiar sales colleagues or email us at sales@gempharmatech.com to obtain detailed data information.
Reference:
[1] Zhang C, et al., Cognitive impairment in Alzheimer's disease FAD4T mouse model: Synaptic loss facilitated by activated microglia via C1qA. Life Sci. 2024 Jan 23;340:122457.
[2] M.S. Gee, et al., A selective p38α/β MAPK inhibitor alleviates neuropathology and cognitive impairment, and modulates microglia function in 5XFAD mouse, Alzheimers Res. Ther. 1 (2020) 45.
[3] L. Tao, et al., Microglia modulation with 1070-nm light attenuates Aβ burden and cognitive impairment in Alzheimer's disease mouse model, Light Sci Appl. 1 (2021) 179.
[4] S. Hong, et al., Complement and microglia mediate early synapse loss in Alzheimer mouse models, Science 6286 (2016) 712–716.
[5] M.J. Vasek, et al., A complement-microglial axis drives synapse loss during virusinduced memory impairment, Nature 7608 (2016) 538–543.
[6] H. Lui, et al., Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation, Cell 4 (2016) 921–935.
[7] D.P. Schafer, et al., Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner, Neuron 4 (2012) 691–705.
[8] B. Stevens, et al., The classical complement cascade mediates CNS synapse elimination, Cell 6 (2007) 1164–1178.
[9] C.J. Bohlen, et al., Microglia in brain development, homeostasis, and neurodegeneration, Annu. Rev. Genet. (2019) 263–288.